SELTENE ERKRANKUNGEN
Phenylketonuria
Phenylketonurie (PKU) ist ein seltener, autosomal-rezessiv vererbter Stoffwechseldefekt von Phenylalanin (Phe). PKU wird durch einen Mangel des Enzyms Phenylalaninhydroxylase (PAH) verursacht, die Phenylalanin in Tyrosin umwandelt. Ein PAH-Mangel führt zur Anreicherung von Phe im Blut und Gehirn.
Carol Buck, Tochter der Nobelpreis- und Pulitzer-Preisträgerin für Literatur Pearl Sydenstricker Buck (26.06.1892–06.03.1973), ist wohl die erste bekannte PKU-Patientin, obgleich ihre Erkrankung damals nicht als solche erkannt wurde. Erst in den 1960er Jahren kam die Bestätigung dafür, dass ihre geistige Behinderung durch PKU bedingt war.

Der norwegische Arzt Dr. Ivar Asbjørn Følling (23.08.1888–24.01.1973) entdeckt PKU nach Untersuchung von Urinproben zweier Kinder mit offenkundiger mentaler Retardierung. Die Mutter hatte einen strengen Geruch des Urins ihres einjährigen Kindes wahrgenommen. Dr. Følling stellt fest, dass der Geruch auf Phenylbrenztraubensäure zurückzuführen war.

Prof. Dr. Horst Bickel (28.06.1918–01.12.2000) veröffentlicht die erste Phenylalanin-arme Diät, nachdem er im Jahr zuvor den Einfluss von Phenylalanin auf die Entstehung der Erkrankung entdeckt hatte. Sheila Jones, ein zweijähriges Mädchen mit schwerer mentaler Retardierung, das nicht stehen, gehen und sprechen konnte, war das erste Kind, das mit einer Phenylalanin-reduzierten Diät behandelt wurde. Unter fortgesetzter Behandlung lernte Sheila zu krabbeln, zu stehen und auf Stühle zu klettern. Sie schlug nicht mehr den Kopf an und hörte auf, ununterbrochen zu weinen.

Horst Bickel, Evelyn Hickmans und John Gerrard werden 1962 mit der John Scott-Medaille für ihre Entdeckung ausgezeichnet. Die John Scott Auszeichnung wird „den verdienstvollsten“ Frauen und Männern verliehen, deren Entdeckungen und Erfindungen auf außerordentliche Weise zum „Komfort, Wohl und Glück“ der Menschheit beigetragen haben. 5 Jahre später wurde diese Auszeichnung Paul M. Zoll für seine Entdeckung des Herzschrittmachers verliehen.
Der US-amerikanische Mikrobiologe Robert Guthrie (28.06.1916–24.06.1995) führt das erste Neugeborenen-Screening auf PKU ein. 1958 wurde bei seiner 15 Monate alten Nichte eine Phenylketonurie diagnostiziert.

Der Schweizer Wissenschaftler Bernhard Schircks beginnt das Studium der organischen Chemie an der Universität Zürich.

Bernhard Schircks schließt seine Promotion im Labor von Professor Viscontini ab, der sich mit der Synthese von Naturstoffen befasste und mit dem Kinderspital Zürich zusammenarbeitete.

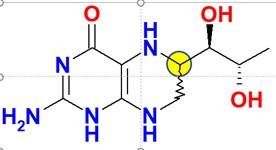
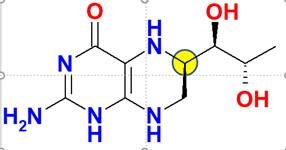
Tetrahydrobiopterin, das Dr. Schircks während seiner Jahre bei Professor Viscontini synthetisiert hatte, wird das erste Mal einer Patientin mit BH4-Mangel verabreicht. Bei der Patientin sank der Phenylalanin-Spiegel innerhalb von wenigen Stunden auf Normalwerte. Die Veröffentlichung dieser Ergebnisse [Archives of disease in childhood (1978), 53(8), 674-6] weckte Hoffnungen und stieß in der Wissenschaftsgemeinde auf große Begeisterung, und Ärzte verschiedener PKU-Kliniken nahmen mit Dr. Schircks Kontakt auf, um sich mit BH4 für ihre Patienten zu versorgen. Das damals verwendete Tetrahydrobiopterin war eine 6R,S-BH4-Mischung, die ca. 30 % des biologisch inaktiven (6S)–Diastereomers enthielt. Es wurde später berichtet, dass dieses Diastereomer sogar eine irreversible Inaktivierung von PAH aus Rattenleber verursacht (Mitchell, J.J., et al., Genet. Med., 2011, 13, 697-707).
Nach einem Postdoc-Jahr in Austin (Texas) gründet Dr. Schircks ein eigenes Unternehmen (Schircks Laboratories) in der Region Zürich und beginnt Tetrahydrobiopterin-Tabletten weltweit auf der Grundlage des Einzelimports von Arzneimitteln zu vertreiben.

Die erste wissenschaftliche Abhandlung berichtet über die Wirksamkeit von BH4 bei einigen PKU-Patienten [Niederwieser A., et al., H. Bickel, U. Wachtel (Eds.), Inherited Diseases of Amino Acid Metabolism, Georg Thieme, Stuttgart, 1985, S. 104-121]. Daraufhin wurden die Nachweise noch zahlreicher und Schircks Laboratories war mit einer wachsenden Nachfrage nach BH4-Tabletten konfrontiert, ohne jemals eine Werbekampagne geführt zu haben.
Hochreines 6R-BH4 wird synthetisiert.
BioMarin beginnt mit der Durchführung einer klinischen Studie, um die Marktzulassung eines Arzneimittels mit der Bezeichnung PhenoptinTM zur Behandlung von Patienten mit BH4-Mangel und PKU zu erhalten.
Das Arzneimittel von BioMarin, das in Kuvan® umbenannt wird, erhält von der US-FDA die Zulassung für die Behandlung der Phenylketonurie bei Patienten, die auf BH4 ansprechen, in Kombination mit einer Phenylalanin-armen Diät.
Kuvan® wird von der EMA zugelassen.
Schircks Laboratories stellt die Herstellung der Sapropterin-Tabletten ein, nachdem Biomarin und Merck Serono den Markt der Tetrahydrobiopterin-Tabletten übernommen haben und auf den Hauptmärkten den Alleinvertrieb für Orphan Drugs erhalten hat.
Dr. Schircks startet die Zusammenarbeit mit Dipharma im Hinblick auf die Entwicklung eines generischen Sapropterin-Arzneimittels.
Diterin® (Sapropterin Dipharma), das erste Generikum von Kuvan®, erhält die Marktzulassung in der Republik Südkorea.
Veröffentlichung der Europäischen Leitlinien für die Optimierung der Phenylketonurie-Therapie.

Sapropterin Dipharma ist in Europa und der Schweiz Verfügbar
Symptomatik
Unbehandelt führen erhöhte Phe-Konzentrationen im Blut und Gehirn zu schwerer, irreversibler geistiger Behinderung, Mikrozephalie, Epilepsie, motorischen Defiziten, ekzematösen Hautausschlägen, Entwicklungsstörungen, Verhaltensauffälligkeiten und psychiatrischen Symptomen. Wenn eine PKU beim Neugeborenen-Screening frühzeitig erkannt und die Behandlung kurz nach der Geburt begonnen wird, kann eine mentale Retardierung verhindert werden: Die Patienten entwickeln allgemeine Fähigkeiten, die unter den breiten Normbereich fallen, erreichen die erwarteten Bildungsstandards und können im Erwachsenenalter ein unabhängiges Leben führen.
Inzidenz in Europa
1 von 10.000 Neugeborenen
Behandlung
Da hohe Phe-Blutkonzentrationen eng mit neurokognitiven Folgen verbunden sind, zielen die bestehenden Behandlungen auf eine Senkung der Phe-Blutkonzentrationen ab. Die Behandlungen schließen eine streng kontrollierte Aufnahme von Phenylalanin (Phe) über die Ernährung in Kombination mit Phe-freien L-Aminosäuren-Präparaten ein sowie eine medikamentöse Therapie, wie z. B. Sapropterindihydrochlorid, das zur Wiederherstellung des Phe-Metabolismus beiträgt.
Die Diät von PKU-Patienten erfordert eine strenge Reduktion oder sogar Elimination von Nahrungsmitteln mit hohem Phenylalanin-Gehalt wie Fleisch, Fisch, Nüsse, Hülsenfrüchte, Käse und andere Milchprodukte.
Stärkehaltige Nahrungsmittel wie Kartoffeln, Brot, Nudeln und Mais sind zu überwachen. Säuglinge können weiterhin gestillt werden, um die vielfältigen Vorzüge der Muttermilch zu nutzen; eventuell ist aber eine Beschränkung der Menge notwendig, in diesem Fall müssen fehlende Nährstoffe ergänzend zugeführt werden. Viele diätetische Nahrungsmittel und diätetische alkoholfreie Getränke, die den Süßstoff Aspartame enthalten, müssen ebenfalls vermieden werden. Heute empfehlen die meisten Ärzte, dass PKU-Patienten ihre Phe-Spiegel ein Leben lang überwachen müssen.
In den Anfangsjahren der PKU-Therapie haben Mütter und Gesundheitsfachkräfte sich oft für das Abstillen entschieden, nachdem beim Baby eine PKU diagnostiziert worden ist. Dies galt damals als die einzig effektive Weise, die Nahrungsaufnahme der Säuglinge zu überwachen und nur die Aufnahme einer genau titrierten und abgemessenen Phe-Menge zu erlauben. Heute wird das Stillen empfohlen und gehört zur üblichen Praxis bei der Versorgung von PKU-Patienten. So wurde auch in einer klinischen Studie nachgewiesen, dass die Gewichtszunahme und die Phe-Serumspiegel im ersten Lebensjahr bei gestillten PKU-Säuglingen günstiger ausfielen als bei nicht gestillten PKU-Säuglingen.
Kose E et al. J Pediatr Nurs. 2018
Van Wegberg AMJ et al. Orphanet J Rare Dis. 2017
 English
English Deutsch
Deutsch