Rare diseases
Phenylketonuria
Phenylketonuria (PKU) is a rare autosomal recessive inborn error of phenylalanine (Phe) metabolism caused by deficiency in the enzyme phenylalanine hydroxylase (PAH) that converts phenylalanine into tyrosine. PAH deficiency leads to accumulation of Phe in the blood and brain.
Carol Buck (daughter of Literature Nobel Prize and Pulizer Prize Pearl Sydenstricker Buck (26/06/1892–06/03/1973) is probably the first known patient affected by PKU, even if at that time the disorder she was suffering from, had not been identified as such. Confirmation that her mental disability was due to PKU came in the 1960’s only.

Norwegian Dr Ivar Asbjørn Følling (23/08/1888-24/01/1973) discovers PKU, analysing urine samples from 2 children with clear mental retardation, whose mother detected strong odour in the urine of her 1-year old child. Dr. Følling discovers that the odour was due to phenylpyruvic acid.

Prof. Dr. Horst Bickel (28/06/1918-01/12/2000) publishes the first diet poor in phenylalanine after having discovered the year before the influence of phenylalanine in the evolution of the disorder. Sheila Jones, a two-year old little girl mentally severely retarded, unable to stand, walk or talk, was the first child treated with a phenylalanine-restricted diet. During her continued treatment Sheila learnt to crawl, to stand and to climb on chairs. She did no longer bang her head and stopped to cry continuously.

Bickel, Eveyn Hickmans and Gerrard win the John Scott Medal in 1962 for their discovery. The John Scott Award is given to “the most deserving” men and women whose inventions have contributed in some outstanding way to the “comfort, welfare and happiness” of mankind. 5 years later, this prize was awarded to Paul M. Zoll, for his discovery of heart pacemaker.
American microbiologist, Robert Guthrie (28/06/1916-24/06/1995) introduces the first newborn screening test for PKU. In 1958, her 15-month-old niece was diagnosed with phenylketonuria.

Swiss scientist Bernard Schircks starts studying organic chemistry at the Zurich University.

Bernard Schircks completes his PhD in the laboratory of Professor Viscontini who was focused at synthesizing natural products and who had collaborations with the Children’s University Hospital in Zurich.

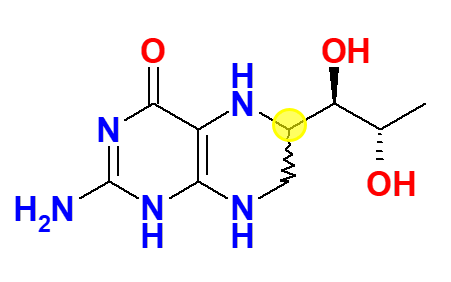
Tetrahydrobiopterin synthesized by Dr Schircks during his years with Professor Viscontini is administered for the first time to a patient affected by BH4 deficiency. The patient saw her phenylalanine levels decreasing within hours to normal levels. Publication of those results (i.e., Archives of disease in childhood (1978), 53(8), 674-6), generated lot of enthusiasm and hope in the scientific community, and physicians of different PKU clinics contacted Dr Schircks to get BH4 for their patients. Tetrahydrobiopterin used in this period was a mixture of 6R,S-BH4, containing about 30% of the biologically inactive 6S diastereomer. Said diastereomer has then been reported to even cause an irreversible inactivation of rat liver PAH (Mitchell, J.J., et al., Genet. Med., 2011, 13, 697-707).
After a one-year post-Doc in Austin (Texas), Dr Schircks sets-up his own Company (Schircks Laboratories) in the region of Zurich, and starts selling on a named patient base, tetrahydrobiopterin tablets around the world.

First scientific paper disclosing BH4 efficacy in some PKU patients (Niederwieser A., et al., H. Bickel, U. Wachtel (Eds.), Inherited Diseases of Amino Acid Metabolism, Georg Thieme, Stuttgart, 1985, pp. 104-121). Then, evidences became even more numerous, and Schircks Laboratories are faced to an increasing demand of BH4 tablets, without having performed any advertising campaigns at all.
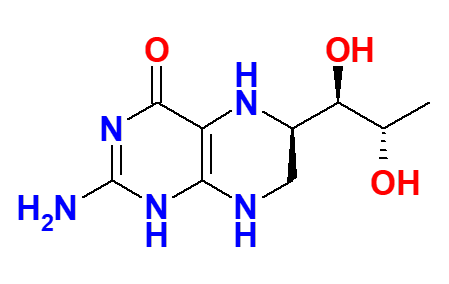
Highly pure 6R-BH4 is synthesized.
BioMarin starts clinical trial aimed at getting marketing authorization of a product called PhenoptinTM for treating BH4 deficient and PKU patients.
BioMarin product, renamed Kuvan®, is approved by US FDA for the treatment of phenylketonuria in BH4-responsive patients together with a restricted diet poor in phenylalanine.
Kuvan® is approved by EMA.
Schircks Laboratories cease the production of sapropterin tablets after Biomarin and Merck Serono took over the tetrahydrobiopterin tablet market and obtained orphan exclusivity in the main markets.
Dr Schircks starts collaborating with Dipharma for the development of a generic sapropterin product
Diterin® (Sapropterin Dipharma), the first generic of Kuvan® is approved in the Republic of South Korea.
Publication of the European guidelines to optimise phenylketonuria care.

Sapropterin Dipharma is available in Europe and Switzerland
Symptoms
If left untreated, increased Phe concentrations in blood and brain cause severe and irreversible intellectual disability, microcephaly, epilepsy, motor deficits, eczematous rash, developmental problems, aberrant behaviour and psychiatric symptoms. PKU early detection during newborn screening and treatment commencement shortly after birth prevent mental retardation: patients fall within the broad normal range of general ability, attain expected educational standards and enable independent lives as adults.
Incidence in Europe
1 Newborn in 10,000.
Treatment
As high blood Phe concentrations are strongly related to neurocognitive outcome, existing treatments aim at decreasing blood Phe concentrations. Treatments include strictly controlled phenylalanine (Phe) amount intakes through diet in combination with Phe-free L-amino acid supplements, and also pharmacological treatments, such as sapropterin dihydrochloride, which helps restoring Phe metabolism.
Diets for PKU patients require severely restricting or even eliminating foods of high phenylalanine content, such as meat, fish, nuts, cheese, legumes, and other dairy products.
Starchy foods such as potatoes, bread, pasta, and corn must be monitored. Infants may still be breastfed to provide all of the benefits of breast milk, but the quantity may need to be limited, resulting in necessary supplementation of missing nutrients. Many diet foods and diet soft drinks containing aspartame sweetener must also be avoided. Today most physicians recommend that PKU patients must keep monitoring their Phe levels throughout their entire life.
In the early years of PKU treatment, mothers and healthcare professionals often decided to discontinue breastfeeding after the diagnosis of PKU in infants. It was believed to be the only effective way to monitor the infant’s intake and allow for precise titration and measurement of the intake of Phe. Today, breastfeeding is encouraged and is a well-established practice in PKU patients care. A clinical study also demonstrated that, in the first year of life, weight gain and serum Phe levels were more favourable in breastfed PKU infants than to non-breastfed PKU infants.
Kose E et al. J Pediatr Nurs. 2018
Van Wegberg AMJ et al. Orphanet J Rare Dis. 2017
 English
English Deutsch
Deutsch