Rare diseases
Niemann Pick C
Niemann–Pick type C is a genetic, neurodegenerative disorder characterised by unique abnormalities of intracellular transport of lipids such as cholesterol in the lysosomes.
Dr. Albert Niemann (23/02/1880-22/03/1921), a German physician, performs the first clear description of what is now known as Niemann-Pick disease (NPD). He completes a clinical report of the infant “Irene D.”, who died at 18 months of age, having been ill all her life. At autopsy, the liver, spleen and lymph node appear enlarged, yellow, fatty and mostly replaced by large vacuolated cells that he wisely believes to be similar to those in Gaucher’s disease.

Dr. Ludwig Pick (31/08/1868-3/02/1944), a German pathologist, starts to accurately describe the pathology of the disease in a series of papers. He identifies the so-called Pick’s cell, a cell type uniquely found in the spleen and bone marrow of NPD patients. Pick’s cells are similar in appearance to “Gaucher’s cells”, however the cytoplasm of these cells appears foamy.

Dr. Klenk demonstrates that NPD is characterized by the pathological accumulation of sphingomyelin in the reticular and histiocytic elements of the liver, spleen and lymph nodes.
C. Crocker and S. Farber publish an extensive review of 18 NPD patients and demonstrate the existence of wide variability both in the age of onset and clinical expression, as in the level of sphingomyelin overload.

Crocker and Farber propose a classification in 4 NPD subgroups:
- Type A (NPA) is characterized by early and severe neurological damage, and a massive visceral and cerebral accumulation of sphingomyelin;
- Type B (NPB) corresponds to a chronic form with marked visceral damage but spares the nervous system;
Types C and D (NPC, NPD) are characterized by neurological and/or neurovisceral damages which could be subacute to moderate; the designation “type D” only relates to a group of patients from Nova Scotia, otherwise similar to type C.
Dr. Roscoe Brady and his group demonstrate a deep deficit in sphingomyelinase enzyme activity in NPD Type A and B patients, but not in Types C/D.

The observation by Dr. Pentchev and collaborators of an abnormal intracellular cholesterol homeostasis in a mutant mouse putative model of NPC leads to a similar study on skin fibroblasts of patients. Dr Pentchev’s group shows that, in NPC cells, the kinetics of cholesterol ester formation induced by low density lipoprotein (LDL) is dramatically slowed down and that an abnormal amount of unesterified cholesterol accumulates in lysosomal vesicles. Later works by Dr. Pentchev and other laboratories bring to the understanding that, in NPC patients, sphingomyelin overload is actually secondary to that of cholesterol. NPC disease begins to be considered as a separate entity and reclassified as a pathology of the intracellular lipid traffic.
Development of NPC first reliable diagnostic tests, applicable to cultured skin fibroblasts. Clinical heterogeneity within type C is asserted, with the recognition of neonatal presentations, rapidly fatal neonatal cholestatic form and forms with early neurological onset. This test which involves the reaction of unesterified cholesterol with fluorescent antibiotic filipin is known as the filipin test and had been first proposed 4 years before.
The demonstration of heterogeneity in NPC patients’ severity of intracellular cholesterol traffic suggests the possible existence of several genes involved in the disease.
After having been hypothesized in 1994, NPC1 gene is identified.
A second gene still coding for transport proteins is identified and named NPC2. It possesses different properties than its analogue NPC1, but both are involved in the same metabolic pathway.
Data from animal studies show that the small molecule miglustat has specific properties that enable its wide tissue distribution and even to cross the blood–brain barrier. Miglustat acts as a competitive inhibitor of the enzyme glucosylceramide synthase, resulting in a reduction of pathological intracellular lipid storage.

Following positive results from preclinical studies, a prospective, randomized clinical trial is designed and implemented to assess the efficacy, safety and tolerability of 12-month treatment with miglustat in juvenile or adult NPC patients (aged 12 years or older). Clinical study results demonstrate some statistically significant improvement in patients under miglustat treatment versus those receiving the standard of care (treatments aimed at relieving disease symptoms).
Long-term data from 12-month extension of miglustat assumption confirm the long-term safety and efficacy of miglustat therapy in stabilizing neurological manifestations in pediatric, juvenile and adult patients.
Identification of NPC1 and NPC2 proteins roles in cholesterol efflux from lysosomes. Cholesterol released from LDL first binds to a specific site on NPC2, which transfers it to a binding site located on NPC1. NPC cells with mutations in NPC1 and/or NPC2 give rise to severe abnormalities in the intracellular transport of lipid, notably cholesterol, glycosphingolipids and sphingosine.
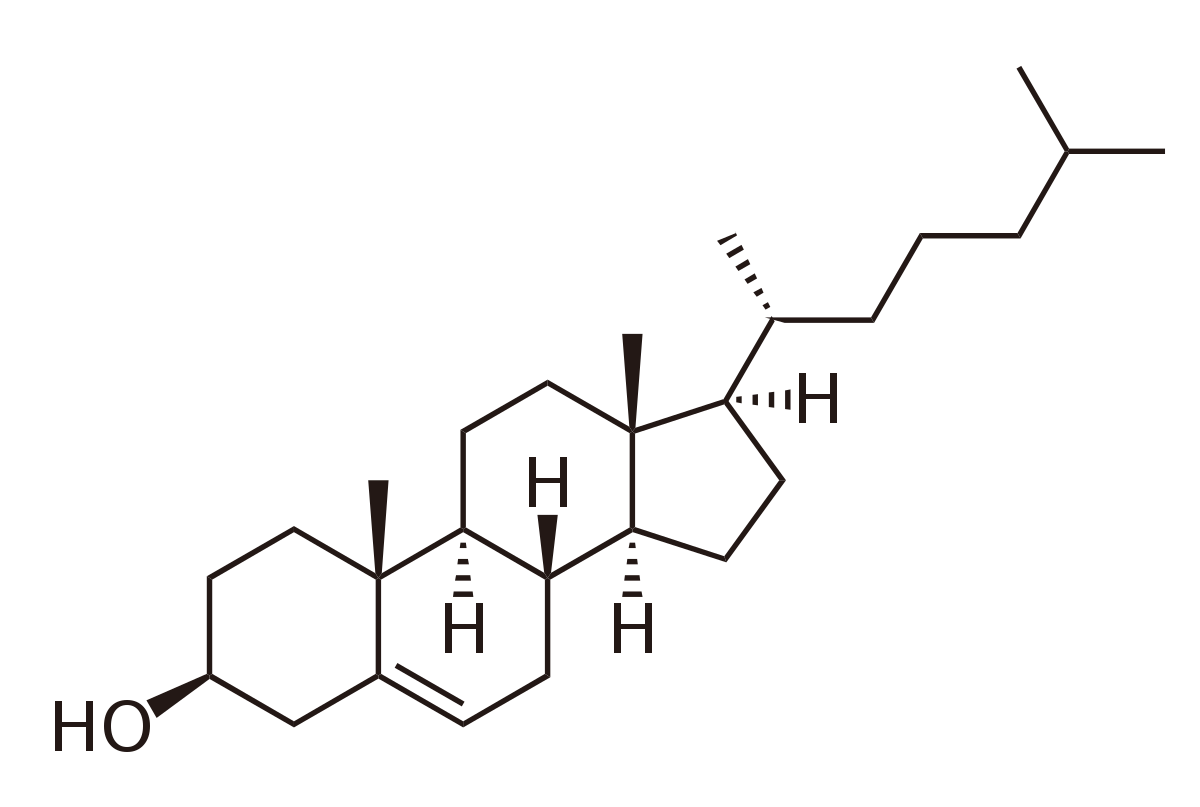
The Swiss company Actelion Pharmaceuticals obtains EMA’s approval for Zavesca® (miglustat 100 mg capsules) for the treatment of progressive neurological manifestations in adult patients and pediatric patients with Niemann-Pick type C disease.
Symptoms
The clinical spectrum of Niemann-Pick type C ranges from a rapidly progressive neonatal form to a slowly progressive adult-onset of neurodegenerative syndrome. Patients can usually survive to their seventh decade.. The early onset form usually also presents with cholestatic jaundice, hepatosplenomegaly and/or acute liver failure. These features may all be absent in the late-onset form. Other common features are: gelastic cataplexy, vertical supranuclear gaze palsy (VSGP), ataxia, dystonia and dementia.
Incidence
1 person in 100,000.
Treatment
Although there is no specific cure for this condition, several clinical trials have identified Miglustat, a substrate reduction therapy, as a treatment option. In some patients, the drug halts or attenuates disease progression.
Lad M et al. Pract Neurol. 2019 Oct;19(5):420-423.
 English
English Deutsch
Deutsch