Rare diseases
Gaucher Disease
Gaucher disease is a rare, autosomal recessive genetic disorder. It is caused by a deficiency of the lysosomal enzyme glucocerebrosidase (also known as acid β-glucosidase), which leads to an accumulation of its substrate, glucosylceramide. This process can lead to splenomegaly and hepatomegaly (spleen and liver enlargement), and bone lesions.
First description of the disease by the medical student Philippe Charles Ernest Gaucher (26/07/1854-25/01/1918), who discovers it in a 32-year-old woman who had an enlarged spleen. A post-mortem exam reveals that cells in the spleen are themselves enlarged. Gaucher described these clinical and pathological findings in his doctoral thesis.

Dr. H. Lieb finds that the chemical cause of the enlarged spleen and liver is the accumulation of sphingoglycolipids in cells, that he erroneously identifies to be cerebroside. However, the optical rotation of the aqueous cleavage product was incompatible with that deduction.

The French chemist, Dr. A. Aghion demonstrates that the accumulating material is glucocerebroside.

Identification of three different Gaucher disease phenotypes, according to the age of symptoms onset: adult form with no impairment of central nervous system (type 1), infantile form, with acute neuronopathic impairment (type 2) and juvenile or chronic neuronopathic form (type 3).
Dr. Roscoe Brady (11/10/1923-13/06/2016) and his group show that people with Gaucher disease make the lipid normally but do not make enough of the enzyme “glucocerebrosidase” to break it down and clear it out of the body. The finding that all patients have some detectable enzyme, later proved to be extremely important in attempts to treat them. Dr. Brady is also remembered for his immense accomplishments in the identification of defects in Niemann-Pick disease, Fabry disease, and Tay-Sachs disease.

Dr. Brady suggests a therapy for Gaucher disease based on replacing the lacking enzyme (ERT approach). Using human placentas, Dr. Peter Pentchev of Dr. Brady’s team isolates a tiny sample of purified glucocerebrosidase.
Dr. Brady’s group devises a test based on the enzyme’s activity to identify people that could develop Gaucher disease, as well as a procedure for prenatal diagnosis.
Two patients receive the purified glucocerebrosidase enzyme through intravenous injection. Thanks to the good biochemical results, Dr. Brady decides to develop a procedure to obtain larger quantities of the enzyme for conducting further clinical trials.
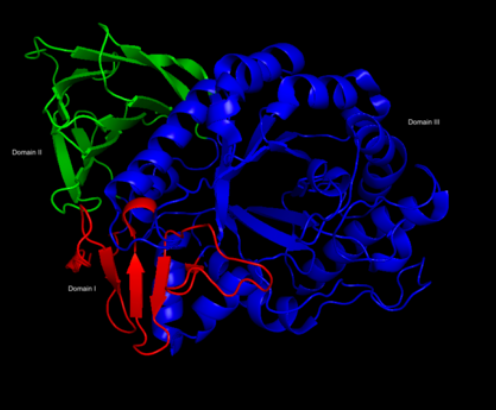
A large-scale purification method is completed, but this preparation of the enzyme produces inconsistent results in clinical trials due to insufficient delivery of the enzyme to the main affected target cells, called macrophages.
Dr. Shoji Tsuji and his team discover the first gene mutation that causes Gaucher disease, which turns out to be an autosomal recessive genetic disease. This means that both the glucocerebrosidase genes a person inherits – one from the mother and one from the father — must be mutated for the person to develop the disease. Dr. Shoji Tsuji will be awarded in 2015 the medal for scientific achievement in Neurology, notably for the great achievements obtained in the elucidation of Gaucher disease.

Dr. Norman Radin hypothesizes a new therapeutical approach to reduce glucocerebroside accumulation through the inhibition of its biosynthesis pathway. This potential treatment approach is defined as substrate reduction therapy (SRT).

A procedure initially sought in 1984 is fine-tuned. It greatly enhances the delivery of glucerebrosidase to macrophages, leading to new clinical trials on Type 1 Gaucher disease patients. The obtained results demonstrate significant clinical benefits, with great reduction in the size of liver and spleen and reduction in bone damage. As a limitation, high amount of enzyme per kg of body weight are however required.
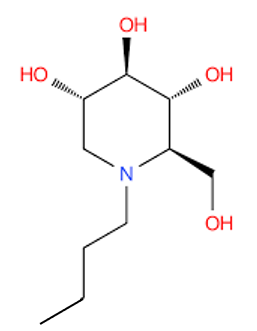
Identification of the glucose analogue, N-butyl-deoxynojirimycin (NB-DNJ, Miglustat), an inhibitor of the enzyme ceramide-specific glucosyltransferase (CSG), which is involved in the first step of glycosphingolipids biosynthesis, including glucorebroside. NB-DNJ was initially developed as an antiviral agent against certain diseases among which HIV.
New ERT clinical trials on type 2 and type 3 Gaucher disease patients show remarkable improvement in treating the systemic manifestation of the disease, but the effect of the intervention on the neurological manifestation remains problematic, because the enzyme difficulty crosses the blood-brain barrier. This is a very impactful limitation of ERT treatments, which together with related high costs, even reduce the possibility to treat the most possible patients in need.
One-year open-label trial to study the effect of miglustat in type 1 Gaucher disease is completed. Results of the study demonstrate a 19% reduction in liver and spleen volumes.
The Swiss company Actelion Pharmaceuticals obtains EMEA’s approval for Zavesca® (miglustat 100 mg capsules), the first oral treatment for type 1 Gaucher disease. Miglustat works as a SRT, preventing the formation of glucocerebroside. Zavesca® is a treatment option for patients for whom enzyme replacement therapy is not amenable due to various reasons, such as allergy and hypersensitivity.
Zavesca® is approved by US FDA.
An open-label, multicenter Phase 3 trial to study the long-term safety and efficacy of Zavesca® in adults with type 1 Gaucher disease is completed. The results show that treatment with miglustat permits to maintain liver size overall.
Symptoms
Gaucher disease is characterized by hepatosplenomegaly, cytopenia, sometimes by severe bone involvement and, in certain forms, by neurological impairment.
The most common form of the disease (Type-1 GD), usually distinguished by the absence of neurological impairment, has a variable clinical presentation, ranging from asymptomatic throughout entire life to early-onset forms presenting in childhood.
Splenomegaly is observed in more than 90% of patients and is sometimes massive, with a spleen weighing up to several kilograms and causing abdominal pain or distension. Indeed, it may be the only clinical sign. Spleen enlargement increases the risk of splenic rupture, but this only occurs very rarely.
Hepatomegaly is noted in 60%–80% of patients. Liver rupture may also be observed and manifests with acute pseudo-surgical abdominal pain.
Bone involvement causes acute pain manifesting as very painful bone crises, predominantly in the pelvis and lower limbs (more rarely in the upper limbs), and/or as chronic pain.
Incidence
In the general population, its incidence is approximately 1/60,000 to 1/40,000 births, rising to 1/800 in Ashkenazi Jews ethnic population.
Treatment
The goal is to treat patients before the onset of complications, the sequelae of which are disabling and irreversible and include massive fibrous splenomegaly, vascular necrosis, secondary osteoarthritis, vertebral compression and fractures, hepatic fibrosis and lung fibrosis.
There are currently two specific types of treatment for GD: enzyme replacement therapy (ERT), which supplies the glucocerebrosidase lacking in the cells, and substrate reduction therapy (SRT), that reduces excess cell glucosylceramide by decreasing its production. Miglustat is a glucosylceramide synthase inhibitor which reduces its biosynthesis.
Stirnemann J et al. Int J Mol Sci. 2017
 English
English Deutsch
Deutsch